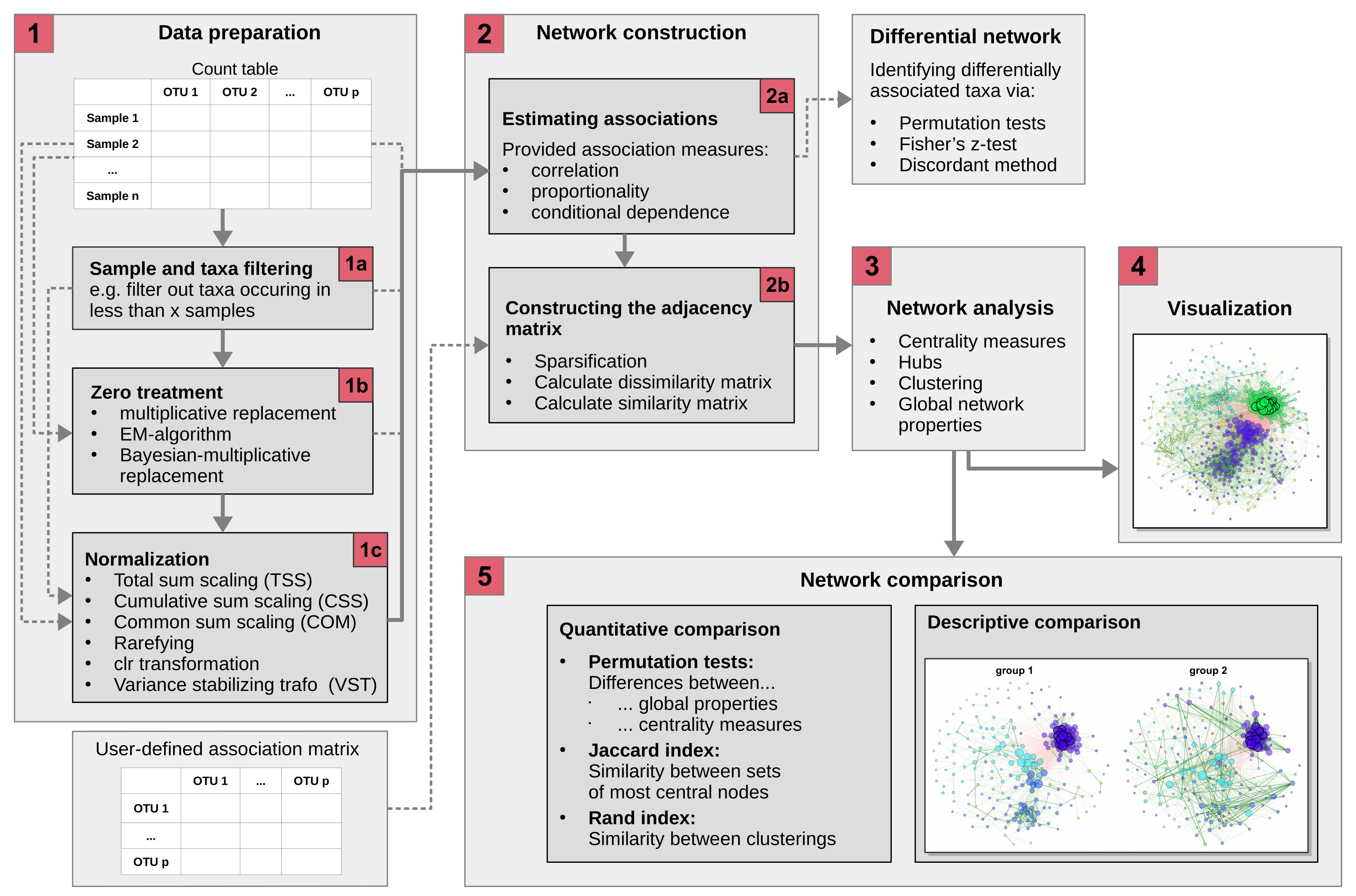
NetCoMi (Network Construction and Comparison for Microbiome Data) is an R package designed to facilitate the construction, analysis, and comparison of networks tailored to microbial compositional data. It implements a comprehensive workflow introduced in Peschel et al. (2020), which guides users through each step of network generation and analysis with a strong emphasis on reproducibility and computational efficiency.
With NetCoMi, users can construct microbial association or dissimilarity networks directly from sequencing data, typically provided as a read count matrix. The package includes a broad selection of methods for handling zeros, normalizing data, computing associations between microbial taxa, and sparsifying the resulting matrices. By offering these components in a modular format, NetCoMi allows users to tailor the workflow to their specific research needs, creating highly customizable microbial networks.
The package supports both the construction, analysis, and visualization of a single network and the comparison of two networks through graphical and quantitative approaches, including statistical testing. Additionally, NetCoMi offers the capability of constructing differential networks, where only differentially associated taxa are connected.
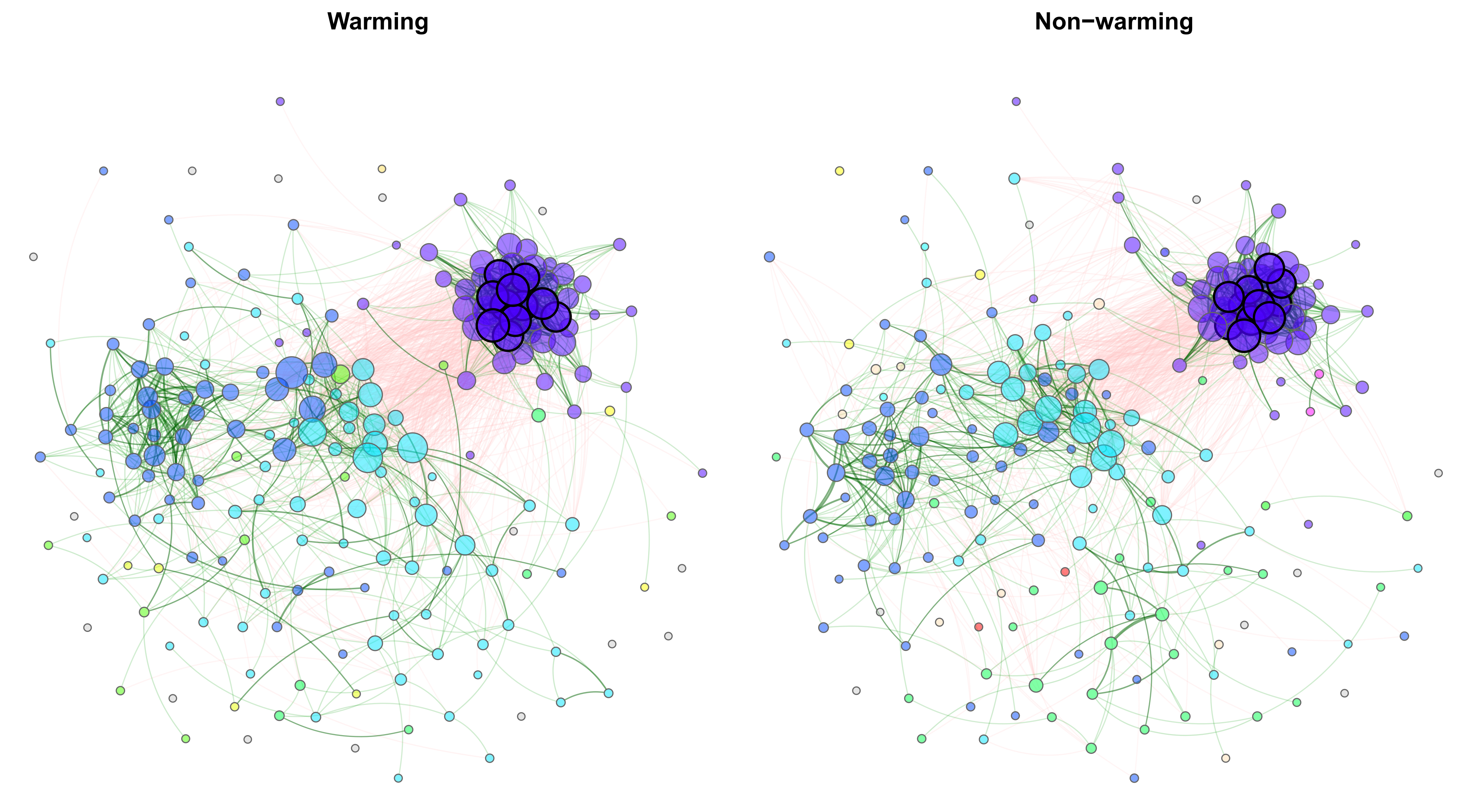
Exemplary network comparison using soil microbiome data (‘soilrep’ data from phyloseq package). Microbial associations are compared between the two experimantal settings ‘warming’ and ‘non-warming’ using the same layout in both groups.
Methods included in NetCoMi
Here is an overview of methods available for network construction, together with some information on their implementation in R:
Association measures:
- Pearson coefficient (
cor()fromstatspackage) - Spearman coefficient (
cor()fromstatspackage) - Biweight Midcorrelation
bicor()fromWGCNApackage - SparCC (
sparcc()fromSpiecEasipackage) - CCLasso (R code on GitHub)
- CCREPE (
ccrepepackage) - SpiecEasi (
SpiecEasipackage) - SPRING (
SPRINGpackage) - gCoda (R code on GitHub)
- propr (
proprpackage)
Dissimilarity measures:
- Euclidean distance (
vegdist()fromveganpackage) - Bray-Curtis dissimilarity (
vegdist()fromveganpackage) - Kullback-Leibler divergence (KLD) (
KLD()fromLaplacesDemonpackage) - Jeffrey divergence (own code using
KLD()fromLaplacesDemonpackage) - Jensen-Shannon divergence (own code using
KLD()fromLaplacesDemonpackage) - Compositional KLD (own implementation following Martin-Fernández et al. (1999))
- Aitchison distance (
vegdist()andclr()fromSpiecEasipackage)
Methods for zero replacement:
- Add a predefined pseudo count to the count table
- Replace only zeros in the count table by a predefined pseudo count (ratios between non-zero values are preserved)
- Multiplicative replacement (
multReplfromzCompositionspackage) - Modified EM alr-algorithm (
lrEMfromzCompositionspackage) - Bayesian-multiplicative replacement (
cmultReplfromzCompositionspackage)
Normalization methods:
- Total Sum Scaling (TSS) (own implementation)
- Cumulative Sum Scaling (CSS) (
cumNormMatfrommetagenomeSeqpackage) - Common Sum Scaling (COM) (own implementation)
- Rarefying (
rrarefyfromveganpackage) - Variance Stabilizing Transformation (VST) (
varianceStabilizingTransformationfromDESeq2package) - Centered log-ratio (clr) transformation (
clr()fromSpiecEasipackage))
TSS, CSS, COM, VST, and the clr transformation are described in (Badri et al. 2020).
Installation
# Required packages
install.packages("devtools")
install.packages("BiocManager")
# Since two of NetCoMi's dependencies are only available on GitHub,
# it is recommended to install them first:
devtools::install_github("zdk123/SpiecEasi")
devtools::install_github("GraceYoon/SPRING")
# Install NetCoMi
devtools::install_github("stefpeschel/NetCoMi",
repos = c("https://cloud.r-project.org/",
BiocManager::repositories()))If there are any errors during installation, please install the missing dependencies manually.
Packages that are optionally required in certain settings are not installed together with NetCoMi. These can be installed automatically using:
If not installed via installNetCoMiPacks(), the required package is installed by the respective NetCoMi function when needed.
Bioconda
Thanks to daydream-boost, NetCoMi can also be installed from conda bioconda channel with
Development version
Everyone who wants to use new features not included in any releases is invited to install NetCoMi’s development version:
devtools::install_github("stefpeschel/NetCoMi",
ref = "develop",
repos = c("https://cloud.r-project.org/",
BiocManager::repositories()))Please check the NEWS document for features implemented on develop branch.